It’s no longer a childhood illness.
Takeaways:
- The Cystic Fibrosis Foundation Patient Data Registry shows that more than 54% of people with cystic fibrosis live 18 years and beyond.
- Longer life expectancy is due to improvements in prescribed therapies and clinical practices.
- Nurses should be acquainted with the underlying cause, treatments, and basic cystic fibrosis care guidelines.
Cystic fibrosis (CF) is the most common progressive, life-shortening, genetic disease among whites. It affects more than 30,000 people in the United States and about 70,000 worldwide. Since 2010, newborn screening for CF has been required in the United States; more than 75% of children with CF are diagnosed by age 2. In the 1950s, children with CF weren’t expected to live beyond 5 years, but the Cystic Fibrosis Foundation Patient Data Registry (CFFPR) shows that more than 54% of people with CF now live 18 years or longer. This percentage has been increasing by nearly 1% annually, and it’s estimated that in 10 years, 70% of Americans with CF will be adults. (See Living with CF.) Based on 2018 CFFPR data, the predicted median age of survival stands at 47.4 years, which means CF should no longer be considered a childhood illness, but rather a lifelong disease diagnosed in infancy. Longer life spans for these patients is the result of improvements in clinical practices and prescribed therapies.
With the growing number of adults with CF and continued improvements in clinical care and prescribed therapies, nurses should be familiar with the underlying cause of CF, its treatments and basic care guidelines, and the changing face of the disease.
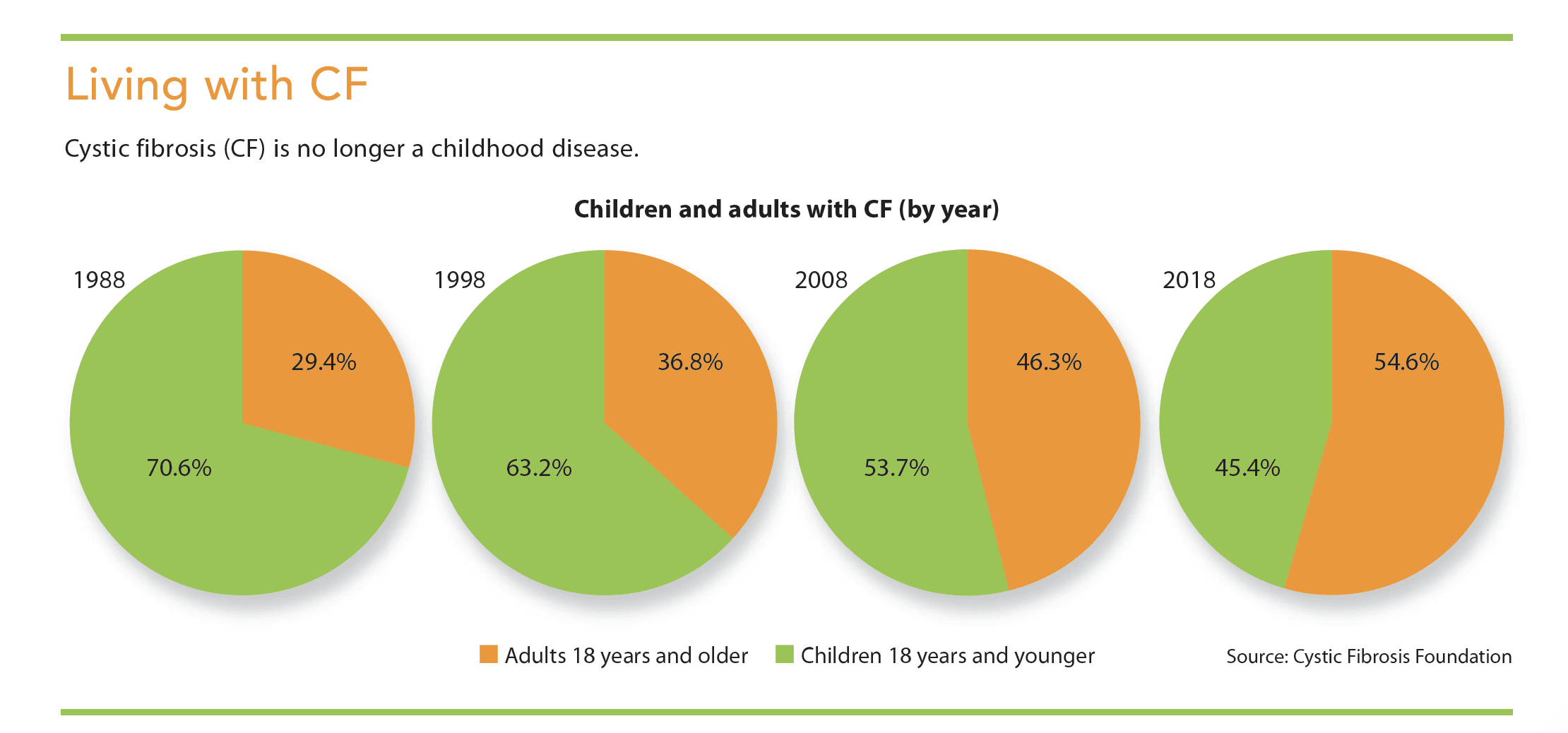
CF explained
Individuals with CF inherit one copy of a genetic mutation of the CF transmembrane conductance regulator (CFTR) from each parent. These mutations result in either a malformed CFTR protein or no CFTR protein being made, which inhibits the movement of chloride in and out of the cells and causes the mucus that lines the body, most notably in the airways, to be thick and sticky.
Since the CFTR gene was identified in 1989, more than 2,000 CFTR mutations have been identified in the Clinical and Functional Translation of CFTR database; 10% to 15% of those mutations have been classified as disease causing. Each mutation leads to diverse symptoms, making CF a multisystem disease. Although CF primarily affects the lungs and pancreas, it also may affect the sinuses, liver, gallbladder, and endocrine and reproductive systems. Mucus may clog lungs, leading to life-threatening infections. Obstruction in the pancreas prevents the release of natural enzymes to break down food for nutrient absorption, leading to steatorrhea, poor weight gain, and slow growth.
The complex, progressive nature of CF requires specialized care by interprofessional clinical teams in a national network of over 130 care centers accredited by the Cystic Fibrosis Foundation (CFF). The care center staff obtain consent to share clinical information through the CFFPR. Since the 1960s, the data collected has been critical to driving care quality improvements, developing clinical guidelines, conducting observational research, and informing the design of clinical trials and ongoing safety studies.
Treatment
Disease and subsequent treatment burden vary widely among CF patients, largely based on age and genotype. This variability is expected to increase with access to CFTR modulators, a new class of Food and Drug Administration (FDA)–approved drugs.
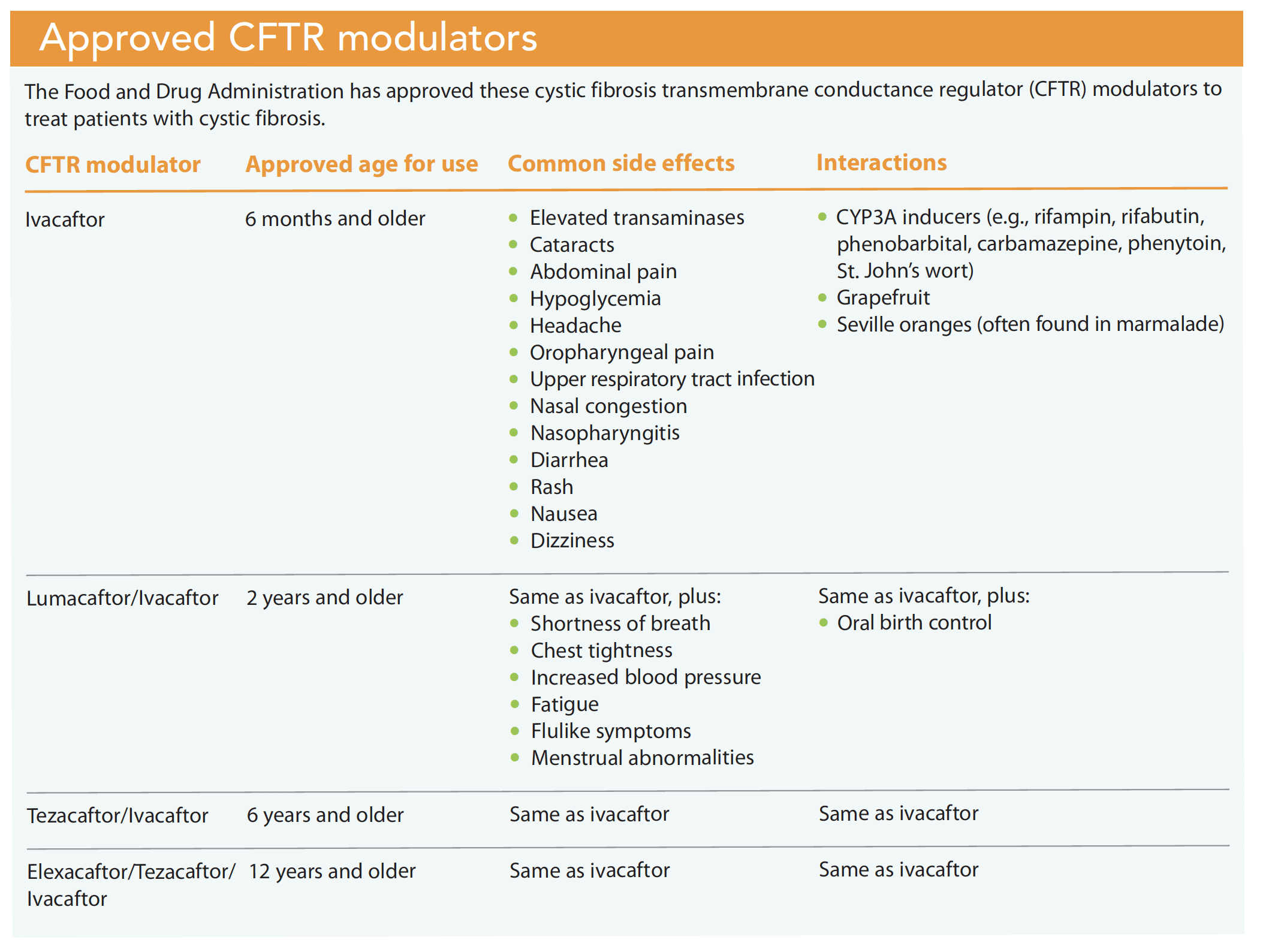
Currently, individuals with CF are required to take many medications to maintain their health and slow disease progression. Pancreatic insufficiency is treated with pancreatic enzyme therapy. Registry data suggest that almost 85% of patients with CF require oral pancreatic enzyme replacement therapy with all meals and snacks. Lung disease is treated with inhaled and oral antibiotics, airway clearance therapy, and mucolytics. Lung treatments are both labor intensive and time-consuming, lasting 2 to 3 hours per day (more if the patient is acutely ill). Although these standard CF therapies address the symptoms of CFTR dysfunction, CFTR modulators (ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor) treat the underlying cause of CF and restore partial function in the gene mutation. (See Approved CFTR modulators.)
Ivacaftor
In 2012, ivacaftor was the first drug in this class to be approved to treat patients with the G551D mutation. It works by keeping the chloride ion channel open. Subjects in clinical trials experienced more than a 10% improvement in their lung function. Unfortunately, only 4.4% of individuals represented in the CFFPR have this genetic mutation.
Lumacaftor/ivacaftor
F508del is the most common CFTR mutation in individuals with CF. According to the CFFPR, approximately 44% of people with CF in the United States are homozygous (they have two copies of this mutation); 40.5% are heterozygous (they have one copy of F508del and another disease-causing mutation). F508del causes the CFTR protein to fold improperly, which prevents it from moving to the cell surface. Because ivacaftor doesn’t affect this protein expression of the CFTR mutation, lumacaftor/ivacaftor was developed. Clinical trials of this medication showed modest (about 3%) overall lung function improvement. Unfortunately, not all individuals with eligible mutations tolerate lumacaftor/ivacaftor because of respiratory and GI side effects. In addition, significant drug-to-drug interactions exist.
Tezacaftor/ivacaftor
Tezacaftor is similar to lumacaftor and is a broad-acting CFTR modulator, resulting in more protein at the cell surface and improved chloride transport. Another benefit of tezacaftor over lumacaftor is that it doesn’t interfere with oral contraceptives. When combined with ivacaftor, tezacaftor/ivacaftor showed a 3.4% to 6.8% improvement in lung function as measured by forced expiratory volume. Lumacaftor’s respiratory side effects, such as chest tightness, weren’t observed.
Elexacaftor/tezacaftor/ivacaftor
Results show that lumacaftor/ivacaftor and tezacaftor/ivacaftor haven’t been as effective as ivacaftor in patients with the G551D mutation. In October 2019, the FDA approved a triple combination therapy that combines ivacaftor and tezacaftor with a third “next-generation” CFTR modulator called elexacaftor. In clinical trials, this triple combination has been highly effective for individuals homozygous and heterozygous for the F508del mutation; they experienced nearly 10% to 14% improvement in their lung function. Highly effective triple combination modulator therapies may allow CF patients to live longer, and standard treatment plans will need to be re-evaluated.
CFTR modulator limitations
Even with effective modulator therapies, the damage that patients with CF have already sustained remains irreversible, leaving a generation of adults with the disease dependent on standard therapies such as inhaled and oral antibiotics to treat their symptoms; some with more advanced disease will require a lung transplant. Additionally, 10% of the CF population have two copies of rare CFTR mutations that don’t have an approved modulator therapy.
Nursing implications
When caring for patients with CF, nurses must be aware of drug-to-food and drug-to-drug interactions and treatment side effects. Individualized treatment plans are the standard of care to address specific genetic mutations. And as this patient population survives longer, nurses will encounter them in a variety of adult care settings.
Interactions
Drug-to-food and drug-to-drug interactions can lead to higher levels of CFTR modulator medication in the blood or reduced therapeutic effect. For example, CFTR modulators must be taken with fat-containing foods to improve medication absorption. CYP3A inducers (such as rifampin), which are prescribed to treat nontuberculous mycobacterial (NTM) lung disease in patients with CF, substantially decrease the therapeutic effect of ivacaftor. This drug-to-drug interaction could prevent patients with CF who have NTM lung disease from realizing the benefits of modulator therapy.
Treatment side effects
Side effects of CFTR modulators vary. Because of potential effects on liver function, liver enzymes should be monitored every 3 months after treatment begins and then annually for the duration of treatment. Some cases of noncongenital cataracts have been reported in pediatric patients when initiating ivacaftor, so baseline and follow-up eye exams should be obtained.
Individualized treatment
CFTR modulator therapies require individualized treatment plans based on genetic mutations, patient age, and eligibility. Pediatric nurses should recognize that the treatment regimens that were once standard CF care may soon be evolving. For example, young children who begin CFTR modulator treatment at 6 months old may not require standard airway clearance therapy or experience pulmonary exacerbations that require hospitalization.
Adult care settings
As more patients with CF are seen in adult care settings, nurses will need to learn about the disease and the model of care used to meet patient needs. Adults with the applicable genetic mutation will be eligible for modulator therapy, but the progressive nature of CF may mean they won’t benefit from this treatment; the airway damage they’ve already sustained can’t be reversed. Modulators will likely not eradicate the bacterial colonization in their lungs from microbes such as Pseudomonas aeruginosa, so these patients will still require anti-infectives and other standard CF therapies.
Clinical guidelines
Nurses in specialty areas such as otolaryngology, endocrinology, reproductive health, and gastroenterology may see an increased number of adults with CF and be asked to coordinate care with CF care teams.
The CFF doesn’t have recommended guidelines for patients with CF who are pregnant, but the lumacaftor/ivacaftor package insert states that the drug may substantially decrease the effectiveness of hormonal contraception. It’s still too early to tell how the long-term effects of the new CFTR modulator therapies will alter hormonal contraception or affect pregnancy and fetal and maternal outcomes. However, a 2016 case report published by Kaminski and colleagues described a safe, uncomplicated pregnancy with a healthy fetal outcome in a woman on modulator therapy. We don’t know how many women with CF have had pregnancies while on CFTR modulators; however, the 2018 CFFPR reports that since 1990, the number of pregnancies among woman with CF has steadily increased, in contrast with those among the general U.S. population, which have declined.
The CFF convenes expert committees to evaluate medical evidence and publish recommendations. Guidelines for infection prevention and control, nutrition, diabetes, colon cancer screening, and mental health are available online.
Infection prevention
Due to potential cross-contamination, nurses caring for patients with CF should adhere to the CFF infection prevention guidelines. In healthcare settings, patients with CF should be on contact precautions, wear disposable surgical masks, and remain 6 feet apart from other people in common areas. If more than one person with CF is part of the setting, specific CFF infection prevention strategies should be implemented. (See Infection prevention strategies.)
The Cystic Fibrosis Foundation recommends these infection prevention strategies when caring for patients with cystic fibrosis (CF).
• Collaborate with the organization’s infection prevention department to establish protocols and checklists for standards of practice when caring for more than one patient with CF.
• Ensure disposable surgical masks are available for patients with CF to wear upon entry to the healthcare facility.
• Whenever possible, place patients with CF in an exam room upon arrival to an outpatient facility.
• If caring for more than one CF patient, do not keep them in the waiting area after they check in; call them on their cellphones when an exam room is available.
• Disinfect multiuse items, such as pens, stethoscopes, and tablets, with an Environmental Protection Agency–registered hospital disinfectant before and after use by patients with CF.
• Implement contact precautions. All healthcare professionals should wear gowns and gloves when caring for more than one patient with CF.
• Disinfect high-touch items such as doorknobs and chairs between use by patients with CF.
Nutrition
Optimal weight for height is associated with better lung function. Individuals with CF require 110% to 200% of the recommended energy intake for gender, age, and height to achieve or maintain a body mass index (BMI) of 23 if male and 22 if female. For patients under age 21, BMI should be at or above the 50th percentile on the Centers for Disease Control and Prevention growth chart. Most patients represented in the CFFPR require pancreatic enzyme replacement therapy to aid fat, carbohydrate, and protein digestion. Salt should be used freely because abnormal CFTR causes salt loss through sweat; a typical “healthy” low-fat, low-salt diet isn’t advised.
Diabetes
CF-related diabetes (CFRD) is the most common comorbidity with a prevalence of almost 30% among adults with CF. Patients with CFRD may have no symptoms, and treatment doesn’t include limiting caloric intake; it’s treated primarily with insulin, not diet, exercise, or oral agents.
Colon cancer screening
According to the CFFPR, the onset of colorectal cancer occurs 20 to 30 years earlier for people with CF than the general population. Screening with colonoscopy for patients with CF is recommended at age 40; for those who’ve received a lung transplant, screening should begin at age 30. Bowel preparation for the procedure is more intense because of thick, sticky mucus in the colon. Patients may require three to four washes, with the last wash occurring 4 to 6 hours before the colonoscopy.
Mental health
Patients with CF have various mental health needs. Chronically ill patients with CF and their caregivers exhibit signs of anxiety and depression at two to three times the rate of the general population. This can affect their ability to sustain daily care, physical health, and quality of life. Patients must work hard to overcome the obstacles of CF self-care, so nonjudgmental support is important as providers partner with CF patients to identify goals. Patients attending a CFF-accredited care center should be screened annually for anxiety and depression. If the screen is positive, they should be offered treatment by a mental or behavioral health provider who is aware of clinical practice guidelines and special requirements for individuals with CF.
Providing quality care
Because CF is no longer a pediatric disease, nurses in primary adult care and specialized fields are more likely to encounter patients with it. Nurses should partner with patients’ CF care teams and familiarize themselves with the CF model of care and special requirements, starting with clinical care guidelines. Many resources are available to help implement guidelines and ensure high-quality care.
The authors work at the Cystic Fibrosis Foundation in Bethesda, Maryland. Paula H. Lomas is senior director of clinical communications, and Quynh T. Tran is director of medical and patient education.
References
Cystic Fibrosis Foundation. 2018 Patient Registry Annual Data Report. 2019. cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-Registry-Annual-Data-Report.pdf
Davies JC, Moskowitz SM, Brown C, et al. VX-659–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 2018;379(17):1599-611.
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214-24.
Guimbellot J, Sharma J, Rowe SM. Toward inclusive therapy with CFTR modulators: Progress and challenges. Pediatr Pulmonol. 2017;52(S48):S4-14.
Hadjiliadis D, Khoruts A, Zauber AG, et al. Cystic fibrosis colorectal cancer screening consensus recommendations. Gastroenterology. 2018;154(3):736-45.
Kaminski R, Nazareth D. A successful uncomplicated CF pregnancy while remaining on ivacaftor. J Cyst Fibros. 2016;15(1):133-4.
Knapp EA, Fink AK, Goss CH, et al. The Cystic Fibrosis Foundation patient registry. Design and methods of a national observational disease registry. Ann Am Thorac Soc. 2016;13(7):1173-9.
Moran A, Brunzelle C, Cohen RC, et al. Clinical care guidelines for cystic fibrosis-related diabetes: A position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33(12):2697-708.
Quittner AL, Abbott J, Georgiopoulos AM, et al. International Committee on Mental Health in Cystic Fibrosis: Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus statements for screening and treating depression and anxiety. Thorax. 2016;71(1):26-34.
Ren CL, Morgan RL, Oermann C, et al. Cystic Fibrosis Foundation pulmonary guidelines: Use of cystic fibrosis transmembrane conductance regulator modulator therapy in patients with cystic fibrosis. Ann Am Thorac Soc. 2018;15(3):271-80.
Saiman L, Siegel JD, LiPuma JJ, et al. Infection prevention and control guideline for cystic fibrosis: 2013 update. Infect Control Hosp Epidemiol. 2014;35(S1):S1-67.